Damage to the heart, such as from a heart attack, does not repair itself and is therefore permanent. The repair ability of heart muscle cells is lost soon after birth, when these cells transition to their adult form. Researchers from the Van Rooij group, led by Eva van Rooij, professor of Molecular Cardiology at UMC Utrecht and group leader at the Hubrecht Institute, discovered that the protein ARID1A plays an important role in regulating this transition.
During a heart attack, part of the heart muscle receives insufficient oxygen due to a blockage in the blood supply. The heart muscle cells cannot cope well with this lack of oxygen and may suffer damage or even die. Once dead heart cells do not re-form in the adult heart, so the heart cannot adequately recover after a heart attack or other damage. In contrast, the hearts of newborn babies are better able to repair themselves by forming new heart muscle cells. Researchers from the Van Rooij group wondered how it could be that the cells lose their repair ability over time, and whether it would be possible to re-activate this ability in an adult heart to repair damage.
Researcher Kees Boogerd explains what happens in mammalian heart muscle cells just after birth: “Initially, the heart muscle cells are still able to repair themselves by dividing to form new cells. We call this regeneration. After a few days, however, the cells take on their mature, adult form. They are then optimally specialized for contraction, making the heart beat firmly, but they can no longer divide properly. This is what we call maturation. So the heart muscle cells switch, so to speak, from regeneration to maturation. We wanted to know how this switch is regulated and whether we could intervene in it.”
Through experiments in mice and cultured human heart tissue, the researchers discovered that the protein ARID1A plays an important role in the switchover. “When we increased the amount of this protein, we saw that maturation was faster and the cells divided less. In contrast, if we turned ARID1A off, we got the opposite effect and the cells started dividing more. So the presence of ARID1A flips the switch toward maturation: the cells mature and lose their ability to divide. We also discovered that ARID1A regulates this by inhibiting another protein, YAP1. These discoveries give us a much better understanding of how dividing of heart muscle cells is regulated,” Boogerd says.
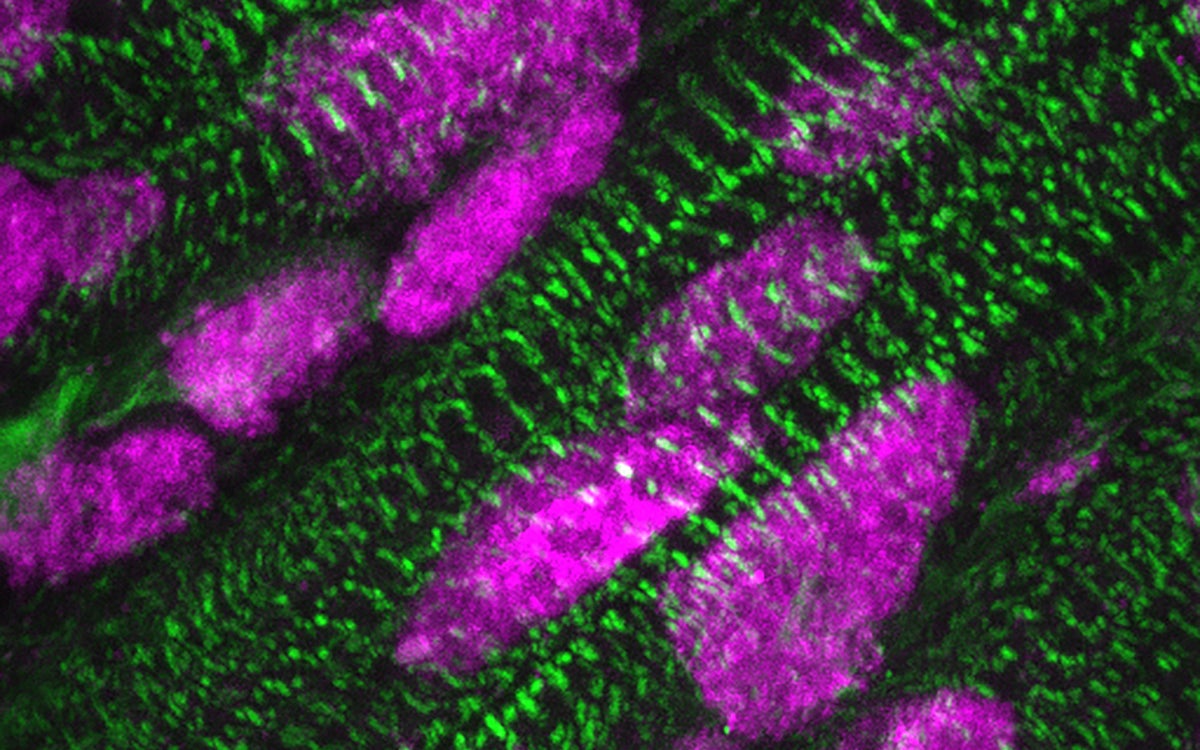
Presence of ARID1A (pink) in the cell nuclei of heart muscle cells (green), a few days after birth. Credit: Kees Boogerd. Copyright: Hubrecht Institute.
With this new knowledge under their belt, the researchers went a step further and investigated whether they could stimulate heart muscle cell regeneration in heart damage. “When we suppressed ARID1A in our model of heart damage, we indeed saw an increase in regeneration. The heart muscle cells around the damaged tissue began to divide. This is fantastic, because it gives us a potential strategy to stimulate heart regeneration. Of course, we still need to investigate this further, to understand even better how to stimulate heart tissue repair, but thanks to our findings we are one step closer to doing so. Hopefully this will allow us to contribute to the development of treatment options for patients with heart damage in the future,” Boogerd concludes.
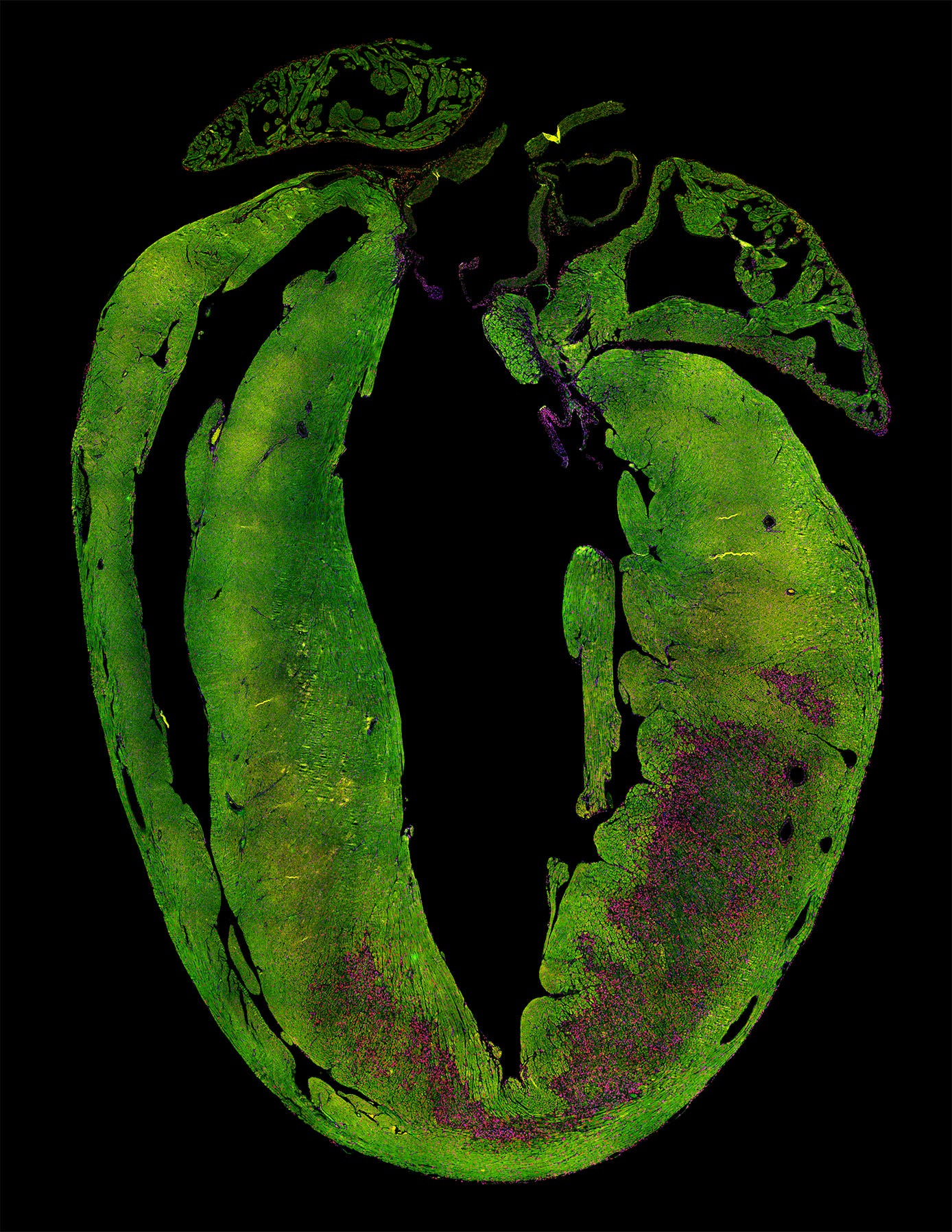
A heart attack leads to loss of heart muscle cells (green) and the formation of scar tissue (purple). This reduces the heart's ability to contract. Credit: Kees Boogerd. Copyright: Hubrecht Institute.
Cardiomyocyte proliferation is suppressed by ARID1A-mediated YAP inhibition during cardiac maturation. Cornelis J. Boogerd, Ilaria Perini, Eirini Kyriakopoulou, Su Ji Han, Phit La, Britt van der Swaan, Jari B. Berkhout, Danielle Versteeg, Jantine Monshouwer-Kloots and Eva van Rooij. Nature Communications, 2023.